Grupo: André Luis Della Valentina, Lucas dos Santos Assmann, Vinícius Bayne Müller
Introdução
A equação de Dirac descreve uma partícula relativística de spin  , como o elétron, com estrutura análoga a da equação de Schrödinger. Ela surgiu inicialmente como tentativa de explicar discrepâncias entre experimentos e teoria para o espectro do átomo de hidrogênio, possibilitando correções para o cálculo da energia do elétron em diferentes níveis (as chamadas correções de estrutura fina). Além disso, por meio dela foi possível prever a existência de antimatéria: descrevendo o elétron, ela também descreve o pósitron.
, como o elétron, com estrutura análoga a da equação de Schrödinger. Ela surgiu inicialmente como tentativa de explicar discrepâncias entre experimentos e teoria para o espectro do átomo de hidrogênio, possibilitando correções para o cálculo da energia do elétron em diferentes níveis (as chamadas correções de estrutura fina). Além disso, por meio dela foi possível prever a existência de antimatéria: descrevendo o elétron, ela também descreve o pósitron.
A fim de compatibilizar a Mecânica Quântica com a Relatividade Especial, a equação diferencial parcial é de primeira ordem tanto no tempo quanto no espaço (diferentemente da equação de Schrödinger, que é de segunda ordem no espaço). A equação de Dirac pode ser escrita de diversas formas; aqui, apresentamo-la explicitamente como um sistema de EDPs acopladas, mais conveniente para os propósitos do trabalho.
Assim como a equação de Schrödinger, a construção da equação de Dirac vem a partir do operador Hamiltoniano, que descreve a evolução temporal do estado quântico em questão:

onde, como anteriormente, os autovalores de  correspondem aos valores possíveis de energia que o sistema pode assumir.
correspondem aos valores possíveis de energia que o sistema pode assumir.
A mudança com relação à Mecânica Quântica não relativística acontece quando consideramos a energia relativística da partícula:

Assim, o Hamiltoniano é modificado para representar a medida da energia relativística total.
Diferentemente da equação de Schrödinger, aqui  não representa apenas uma função de onda, mas sim um conjunto de quatro delas. Usando a notação
não representa apenas uma função de onda, mas sim um conjunto de quatro delas. Usando a notação
 ,
,
as componentes de  representam as funções de onda associadas ao elétron e ao pósitron:
representam as funções de onda associadas ao elétron e ao pósitron:  (
( ) representa a função de onda do elétron com spin up (down), e
) representa a função de onda do elétron com spin up (down), e  (
( ) representa a função de onda do pósitron com spin up (down). O objeto é chamado de spinor.
) representa a função de onda do pósitron com spin up (down). O objeto é chamado de spinor.
Dedução da equação de Dirac em uma e duas dimensões
Consideraremos neste trabalho a equação de Dirac em uma e duas dimensões,  e
e  . A escolha dessas coordenadas se dá pela conveniência do acoplamento das EDPs: nesse caso, as quatro equações acopladas passam a ser acopladas duas a duas, facilitando o estudo do sistema.
. A escolha dessas coordenadas se dá pela conveniência do acoplamento das EDPs: nesse caso, as quatro equações acopladas passam a ser acopladas duas a duas, facilitando o estudo do sistema.
Construção do Hamiltoniano completo
Consideremos uma partícula sob ação de um potencial  (onde
(onde  ), que afeta a energia potencial da partícula, e de um potencial "escalar"
), que afeta a energia potencial da partícula, e de um potencial "escalar"  , que afeta a massa de repouso da mesma. Seguindo uma das propostas possíveis para o Hamiltoniano, temos
, que afeta a massa de repouso da mesma. Seguindo uma das propostas possíveis para o Hamiltoniano, temos

onde  ;
;  e
e  são matrizes 4x4 adimensionais e
são matrizes 4x4 adimensionais e  é o vetor momento linear da partícula.
é o vetor momento linear da partícula.
Pode-se mostrar que  e devem satisfazer certos vínculos, limitando as escolhas possíveis para essas matrizes. A escolha mais simples e usualmente adotada consiste em tomar
e devem satisfazer certos vínculos, limitando as escolhas possíveis para essas matrizes. A escolha mais simples e usualmente adotada consiste em tomar




Sendo  , podemos escrever o produto escalar
, podemos escrever o produto escalar  como
como

Portanto, em notação matricial o Hamiltoniano pode ser escrito como


Unidades naturais e redução para duas dimensões
A fim de simplificar o formalismo, adotamos as chamadas "unidades naturais", onde  . Note que isso equivale a reescalar as quantidades físicas do problema por um fator adequado. Ao fazer
. Note que isso equivale a reescalar as quantidades físicas do problema por um fator adequado. Ao fazer  , também assumimos que a partícula está no limite relativístico.
, também assumimos que a partícula está no limite relativístico.
Além disso, queremos estudar o problema em duas dimensões. Observamos que  ; logo,
; logo,  . Portanto, temos o Hamiltoniano simplificado
. Portanto, temos o Hamiltoniano simplificado

Forma explícita final
Retornando ao problema original, queremos resolver
![{\displaystyle i\hbar {\frac {\partial }{\partial t}}\Psi =H\Psi \to \left[iI_{4}{\frac {\partial }{\partial t}}-H\right]\Psi =0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a47cd71f320c16b7c19d774e965a50d7a9ffe74c)
Novamente utilizando a notação matricial, obtemos

Realizando a multiplicação matricial, pode-se ver que se obtém dois sistemas acoplados: com e com . Escolhendo o sistema de com :

Simplificando e isolando a derivada temporal, obtemos por fim

Por fim, a equação em uma dimensão () é facilmente obtida: basta fazer 

Discretização
A equação de Dirac 1D pode ser escrita, na forma matricial, como
![{\displaystyle i\partial _{t}\mathbf {\Phi } =[-i\sigma _{1}\partial _{x}+\sigma _{3}]\mathbf {\Phi } +[V(t,x)I_{2}]\mathbf {\Phi } }](https://wikimedia.org/api/rest_v1/media/math/render/svg/856d45f8bb00f22f263d8cb7b4beaa0994aca01b)
onde  e
e  é matriz identidade de dimensão 2. As matrizes de Pauli
é matriz identidade de dimensão 2. As matrizes de Pauli  são escritas, aqui, como
são escritas, aqui, como  e
e  .
.
Para discretizar uma equação diferencial parcial como essa, é necessário discretizar o espaço e o tempo. Convenciona-se  como um passo finito de tempo e
como um passo finito de tempo e  como um passo finito no espaço, de tal forma que
como um passo finito no espaço, de tal forma que  , onde
, onde  são números inteiros. Define-se a notação
são números inteiros. Define-se a notação  e também
e também  . Discretiza-se as derivadas parciais explicitamente com uma expansão em série de taylor da própria função:
. Discretiza-se as derivadas parciais explicitamente com uma expansão em série de taylor da própria função:

Considerando uma derivada discretizada  e truncando na primeira ordem:
e truncando na primeira ordem:

O processo é completamente análogo para a derivada espacial, porém para facilitar a aplicação do método mantém-se o espaço centrado em  , em outras palavras faz-se uma expansão em torno de
, em outras palavras faz-se uma expansão em torno de  , obtendo:
, obtendo:

Com isso, obtém-se uma equação para um método explícito no tempo da equação de Dirac 1D.
![{\displaystyle i\delta _{t}\mathbf {\Phi _{j}^{n}} =[-i\sigma _{1}\delta _{x}+\sigma _{3}]\mathbf {\Phi _{j}^{n}} +[V_{j}^{n}I_{2}]\mathbf {\Phi _{j}^{n}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/066376632627056eca4aa2be571668af4d36d9ae)
![{\displaystyle i{\frac {\mathbf {\Phi _{j}^{n+1}} -\mathbf {\Phi _{j}^{n}} }{\Delta t}}=[-i\sigma _{1}\delta _{x}+\sigma _{3}]\mathbf {\Phi _{j}^{n}} +[V_{j}^{n}I_{2}]\mathbf {\Phi _{j}^{n}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/7446ce3f82727c2b53fa903abb6796149e3ae1b4)
![{\displaystyle i\mathbf {\Phi _{j}^{n+1}} =\mathbf {\Phi _{j}^{n}} +\Delta t[-i\sigma _{1}\delta _{x}+\sigma _{3}]\mathbf {\Phi _{j}^{n}} +\Delta t[V_{j}^{n}I_{2}]\mathbf {\Phi _{j}^{n}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/ab7fbc7e09a9758938eb89013fe5030a54e5abc1)
Pode-se também desenvolver um método implícito no tempo fazendo a expansão de  em torno de
em torno de  , obtendo:
, obtendo:

Ao aplicar esta aproximação na equação discretizada basta dar um passo a frente em todos os elementos, obtendo um método implícito no tempo, já que há dependência com  .
.
![{\displaystyle i\mathbf {\Phi _{j}^{n+1}} =\mathbf {\Phi _{j}^{n}} +\Delta t[-i\sigma _{1}\delta _{x}+\sigma _{3}]\mathbf {\Phi _{j}^{n+1}} +\Delta t[V_{j}^{n+1}I_{2}]\mathbf {\Phi _{j}^{n+1}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/315ca5c2ae8c8d289445d2fad96a783defa76791)
Neste trabalho, foram utilizados dois métodos de integração numérica diferentes: o de Crank-Nicolson, para a equação de Dirac em uma dimensão, e o de Leap-Frog, para a equação em duas dimensões.
Método de Crank-Nicolson
O método de Crank-Nicolson (CN) consiste em uma média entre um método explícito e outro implícito no espaço. Utilizar-se-á a notação  para representar justamente a média entre ambos os métodos, ou seja:
para representar justamente a média entre ambos os métodos, ou seja:

Define-se a notação:

Dessa maneira, enuncia-se o método CN para a equação de Dirac 1D como:
![{\displaystyle i\delta _{t}\mathbf {\Phi _{j}^{n}} =[-i\sigma _{1}\delta _{x}+\sigma _{3}]\mathbf {\Phi _{j}^{n+1/2}} +[V_{j}^{n+1/2}I_{2}]\mathbf {\Phi _{j}^{n+1/2}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/31e9be8f4009379093880a6419a1bfc7236ab22d) ,
,
onde  são as discretizações explícitas das derivadas.
são as discretizações explícitas das derivadas.
Para que seja possível aplicar e estudar o método, é necessário passar da notação matricial para escalar:

![{\displaystyle i{\frac {\mathbf {\Phi _{j}^{n+1}} -\mathbf {\Phi _{j}^{n}} }{\Delta t}}=-{\frac {i}{2}}\sigma _{1}\left[{\frac {\mathbf {\Phi _{j+1}^{n+1}} -\mathbf {\Phi _{j-1}^{n+1}} }{2h}}+{\frac {\mathbf {\Phi _{j+1}^{n}} -\mathbf {\Phi _{j-1}^{n}} }{2h}}\right]+\sigma _{3}\left({\frac {\mathbf {\Phi _{j}^{n+1}} +\mathbf {\Phi _{j}^{n}} }{2}}\right)+I_{2}\left({\frac {V_{j}^{n+1}\mathbf {\Phi _{j}^{n+1}} +V_{j}^{n}\mathbf {\Phi _{j}^{n}} }{2}}\right)}](https://wikimedia.org/api/rest_v1/media/math/render/svg/36c63cac7e0404678553d9417eff8e3740d63a6e)
Isolando cada tempo em um lado da igualdade:
![{\displaystyle \left[I_{2}+{\frac {i}{2}}\Delta t\sigma _{3}+{\frac {i}{2}}\Delta tV_{j}^{n+1}I_{2}\right]\mathbf {\Phi _{j}^{n+1}} +{\frac {\Delta t}{4h}}\sigma _{1}\left[\mathbf {\Phi _{j+1}^{n+1}} -\mathbf {\Phi _{j-1}^{n+1}} \right]=}](https://wikimedia.org/api/rest_v1/media/math/render/svg/11db4df7b507157a75b89ad679b87003f3ef6857)
![{\displaystyle \left[I_{2}-{\frac {i}{2}}\Delta t\sigma _{3}-{\frac {i}{2}}\Delta tV_{j}^{n}I_{2}\right]\mathbf {\Phi _{j}^{n}} -{\frac {\Delta t}{4h}}\sigma _{1}\left[\mathbf {\Phi _{j+1}^{n}} -\mathbf {\Phi _{j-1}^{n}} \right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8730f3d4a62979df98a9dba9c63c334ea822a579)
Abrindo as matrizes  e e operando-as sobre o vetor
e e operando-as sobre o vetor  na equação, tem-se:
na equação, tem-se:

Pode-se realizar as operações matriciais e escrever duas equações escalares. Para facilitar a notação, utiliza-se  e
e  :
:
![{\displaystyle {\begin{cases}\left[1+{\frac {i\Delta t}{2}}(V_{j}^{n+1}+1)\right]f_{j}^{n+1}+{\frac {\Delta t}{4h}}(g_{j+1}^{n+1}-g_{j-1}^{n+1})=\left[1-{\frac {i\Delta t}{2}}(V_{j}^{n}+1)\right]f_{j}^{n}-{\frac {\Delta t}{4h}}(g_{j+1}^{n}-g_{j-1}^{n})\\\left[1+{\frac {i\Delta t}{2}}(V_{j}^{n+1}-1)\right]g_{j}^{n+1}+{\frac {\Delta t}{4h}}(f_{j+1}^{n+1}-f_{j-1}^{n+1})=\left[1-{\frac {i\Delta t}{2}}(V_{j}^{n}-1)\right]g_{j}^{n}-{\frac {\Delta t}{4h}}(f_{j+1}^{n}-f_{j-1}^{n})\end{cases}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/124cc692a9582c970eb9016ec040d79fadb3e1cc)
Tem-se então um número  de equações onde é o número de elementos do espaço discretizado. Portanto o primeiro termo das duas equações gera uma matriz diagonal, pois multiplica os termos espaciais dependentes de ; já o segundo termo gera uma matriz tridiagonal com diagonal principal nula. Nota-se que os primeiros termos dos dois lados da igualdade são o conjugado um do outro: define-se, portanto,
de equações onde é o número de elementos do espaço discretizado. Portanto o primeiro termo das duas equações gera uma matriz diagonal, pois multiplica os termos espaciais dependentes de ; já o segundo termo gera uma matriz tridiagonal com diagonal principal nula. Nota-se que os primeiros termos dos dois lados da igualdade são o conjugado um do outro: define-se, portanto,  e
e  .
.

Considerando que o potencial  é só função da posição, escreve-se o método como:
é só função da posição, escreve-se o método como:
 ,
,
onde
 .
.
Por fim, pode-se escrever o método resolvendo o sistema
 ,
,
onde  ,
,  ,
,  ,
,  ,
,  e
e  .
.
Com isso, e com condições iniciais e de contorno bem estabelecidas, já é possível aplicar o método, dado que todas essas matrizes dependem somente dos parâmetros do sistema.
Estabilidade Crank-Nicolson
Utilizar-se-á o método de von Neumann para analisar a estabilidade do método de Crank-Nicolson para a equação de Dirac unidimensional. Para tanto, supõe-se que a função  pode ser dada pela série de Fourier
pode ser dada pela série de Fourier

Devido à independência linear de cada termo do somatório, ao substituir na equação do método haverá uma equação para cada ente do somatório. Se  , então pode-se dizer que o método estável, já que dessa forma garante-se uma não divergência.
, então pode-se dizer que o método estável, já que dessa forma garante-se uma não divergência.
Aplica-se um termo geral da série de índice  no método CN para a equação de Dirac 1D:
no método CN para a equação de Dirac 1D:
![{\displaystyle \left[I_{2}+{\frac {i}{2}}\Delta t\sigma _{3}+{\frac {i}{2}}\Delta tV_{j}^{n+1}I_{2}\right]\mathbf {A} ^{n+1}e^{ikqjh}+{\frac {\Delta t}{4h}}\sigma _{1}\mathbf {A} ^{n+1}\left[e^{ikq(j+1)h}-e^{ikq(j-1)h}\right]=\left[I_{2}-{\frac {i}{2}}\Delta t\sigma _{3}-{\frac {i}{2}}\Delta tV_{j}^{n}I_{2}\right]\mathbf {A} ^{n}e^{ikqjh}-{\frac {\Delta t}{4h}}\sigma _{1}\mathbf {A} ^{n}\left[e^{ikq(j+1)h}-e^{ikq(j-1)h}\right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/063cae16464b2521ffda10233f3979d7764b70d5)
Divide-se tudo por  e isola-se
e isola-se  :
:
![{\displaystyle \left[I_{2}+{\frac {i}{2}}\Delta t\sigma _{3}+{\frac {i}{2}}\Delta tV_{j}^{n+1}I_{2}+{\frac {\Delta t}{4h}}\sigma _{1}\left(e^{ikqh}-e^{-ikqh}\right)\right]\mathbf {A} ^{n+1}=\left[I_{2}-{\frac {i}{2}}\Delta t\sigma _{3}-{\frac {i}{2}}\Delta tV_{j}^{n}I_{2}-{\frac {\Delta t}{4h}}\sigma _{1}\left(e^{ikqh}-e^{-ikqh}\right)\right]\mathbf {A} ^{n}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5f4ee31dc98c2753dcd52ea1f72ab9fe542b4d6b)
![{\displaystyle \left[I_{2}+{\frac {i}{2}}\Delta t\sigma _{3}+{\frac {i}{2}}\Delta tV_{j}^{n+1}I_{2}+{\frac {\Delta t}{4h}}\sigma _{1}\left({\frac {\sin(kqh)}{2i}}\right)\right]\mathbf {A} ^{n+1}=\left[I_{2}-{\frac {i}{2}}\Delta t\sigma _{3}-{\frac {i}{2}}\Delta tV_{j}^{n}I_{2}-{\frac {\Delta t}{4h}}\sigma _{1}\left({\frac {\sin(kqh)}{2i}}\right)\right]\mathbf {A} ^{n}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/91045c135ca2637beee0fccb3f8f26dc4d4f39cd)
![{\displaystyle \left[I_{2}+{\frac {i}{2}}\Delta t\sigma _{3}+{\frac {i}{2}}\Delta tV_{j}^{n+1}I_{2}-{\frac {i\Delta t}{4h}}\sigma _{1}\left({\frac {\sin(kqh)}{2}}\right)\right]\mathbf {A} ^{n+1}=\left[I_{2}-{\frac {i}{2}}\Delta t\sigma _{3}-{\frac {i}{2}}\Delta tV_{j}^{n}I_{2}+{\frac {i\Delta t}{4h}}\sigma _{1}\left({\frac {\sin(kqh)}{2}}\right)\right]\mathbf {A} ^{n}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e5bcc4e503cf1206d9c6d92fcb89bd4b39fe7db0)
Nota-se que os termos que multiplicam o fator são o conjugado um do outro. Define-se  como a componente l escalarda multiplicação da matriz
como a componente l escalarda multiplicação da matriz  pelo vetor
pelo vetor  :
:

 ,
,
onde  é sempre diferente de zero, visto que a parte real é dada por uma matriz identidade constante.
é sempre diferente de zero, visto que a parte real é dada por uma matriz identidade constante.
Mostra-se, portanto, que a razão entre os coeficientes da série de Fourier nunca diverge, ou seja, o método é incondicionalmente estável.
Simulações em Julia
Equação 1D
Fez-se simulações do método de Crank-Nicolson para a equação de Dirac unidimensional em três configurações de potenciais diferentes: Nulo (Partícula Livre), Poço Infinito e Oscilador Harmônico Simples.
Para todos os casos utilizou-se uma condição inicial de um pacote gaussiano de desvio padrão  para uma das componentes de , sendo a outra componente nula. O módulo quadrado do pacote gaussiano deve ter área unitária dentro da malha utilizada (de
para uma das componentes de , sendo a outra componente nula. O módulo quadrado do pacote gaussiano deve ter área unitária dentro da malha utilizada (de  até
até  ), por isso a constante de normalização deve ser
), por isso a constante de normalização deve ser  .
.
Segue trecho do código comum a todos as simulações realizadas; a única diferença é que em um caso do OHS o pacote gaussiano é deslocado do meio da malha para a posição  .
.
using Plots
using LaTeXStrings
using LinearAlgebra
using Integrals
function init(x, sigma)
arg1 = @. ((-(x-25)^2)/(2*sigma^2))
arg2 = 1/sqrt(sqrt(pi)*sigma)
return arg2*exp.(arg1) ##Inicializo com um pacote gaussiano
end
function OHS(V,x, k)
V = @. 0.5*k*(x-25)^2
return V
end
function poco(V, h)
#Quero colocar nas posições 18 e 32
V[round(Int64, 18/h)] = 100000
V[round(Int64, 32/h)] = 100000
return V
end
##Matrizes do método de Crank-Nicholson
function matriz(dt,h,L, V)
A = Diagonal(ones(L).*(1 .+ im*dt*0.5.*(V.+1)))
B = LinearAlgebra.Tridiagonal(ones(L-1).*(-dt/(4*h)), zeros(L), ones(L-1).*(dt/(4*h)))
C = Diagonal(ones(L).*(1 .+ im*dt*0.5.*(V.-1)))
A = Array(A)
B = Array(B)
C = Array(C)
A_i = inv(A)
B_i = inv(B)
C_i = inv(C)
AiB = A_i*B
CiB = C_i*B
D = B_i*conj(A) + CiB
E = I(L) + C_i*conj(C)
F = B_i*A -CiB
G = A_i*conj(A) + I(L)
H = AiB + B_i*conj(C)
J = AiB - B_i*conj(C)
F_i = inv(F)
J_i = inv(J)
K = F_i*D
L = F_i*E
M = J_i*G
N = J_i*H
return K, L, M, N
end
function CN(size, h, tsize, V, dt, ci)
f = zeros(Complex, tsize, size) #Cada linha representa vetor posição em um tempo diferente. ## phi 1
g = zeros(Complex, tsize, size) ##phi 4
A, B, C, D = matriz(dt, h, size, V) #Matrizes do método Crank-Nicholson
f[1, :] = ci ##Condição Inicial
g[1, :] = zeros(size)
##Condições de Contorno
f[:, 1] = zeros(tsize)
f[:, end] = zeros(tsize)
g[:, 1] = zeros(tsize)
g[:, end] = zeros(tsize)
i=2 #Não interfiro nos contornos
while i<tsize
f[i, :] = A*f[i-1, :] - B*g[i-1, :]
g[i, :] = C*f[i-1, :] - D*g[i-1, :]
i+=1
end
return f, g
end
Partícula Livre
Aplicou-se o código demonstrado com potencial nulo para simular o caso da partícula livre. Segue abaixo código utilizado para gerar a animação:
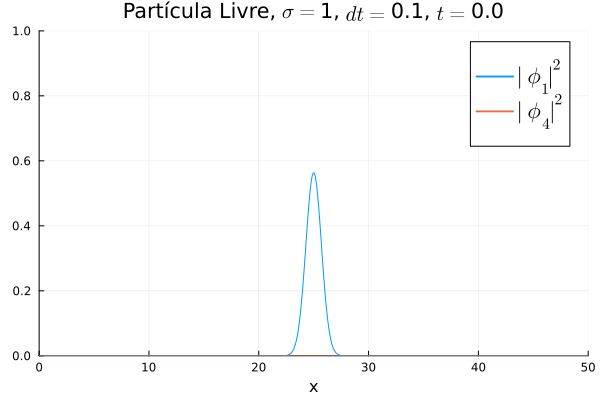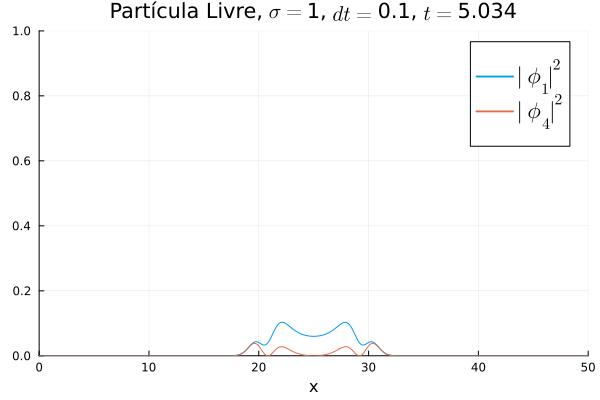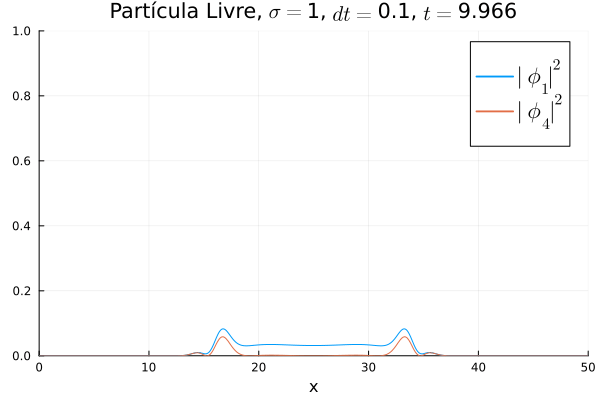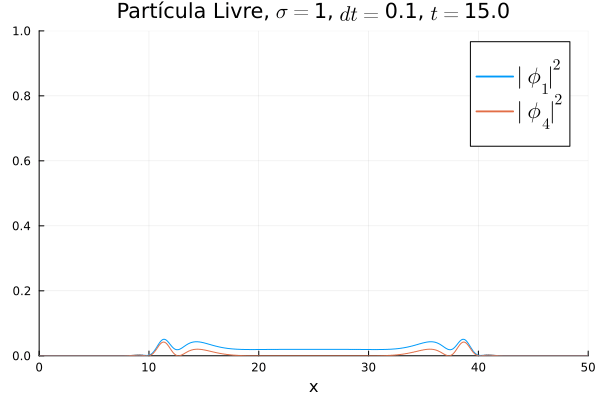
Nota-se que mesmo que  seja nula no início, a existência da partícula
seja nula no início, a existência da partícula  (neste caso, o elétron com spin up) gera a outra (pósitron com spin down). O comportamento é de dispersão, ou seja, a tendência é que a posição fique cada vez menos definida: a partícula livre, por estar fora da ação de um potencial, apresenta momento bem definido; pelo Princípio da Incerteza, a posição torna-se incerta. Nota-se também que as bordas estão longe o suficiente na escala de tempo utilizada, de maneira que as condições de contorno não afetam a simulação.
(neste caso, o elétron com spin up) gera a outra (pósitron com spin down). O comportamento é de dispersão, ou seja, a tendência é que a posição fique cada vez menos definida: a partícula livre, por estar fora da ação de um potencial, apresenta momento bem definido; pelo Princípio da Incerteza, a posição torna-se incerta. Nota-se também que as bordas estão longe o suficiente na escala de tempo utilizada, de maneira que as condições de contorno não afetam a simulação.
function main()
L = 50 #Dimensão espacial matriz
k=0.1 #"Constante Elástica" do oscilador harmônico
sigma = 1 #Largura do pacote gaussiano
dt = 0.02
size= 10*L
h = L/size ##Passo espacial
tmax = 15
tsize = round(Int64, tmax/dt) #Tamanho do vetor tempo
t = LinRange(0, tmax, tsize)
x = LinRange(0, L, size)
V = zeros(size)
xi = init(LinRange(0, L, size), sigma) #Condição inicial pacote gaussiano
f,g =CN(size,h,tsize, V, dt,xi)
i=1
anim = @animate while i<=tsize
f_real = real(f[i, :])
f_imag = imag(f[i, :])
g_real = real(g[i, :])
g_imag = imag(g[i, :])
probf = abs2.(f[i, :])
probg = abs2.(g[i, :])
prob = probf + probg
p=plot(x, probf, label=L"$|\phi_1|^2$", legendfont=font(15))
plot!(x, probg, label=L"$|\phi_4|^2$", legendfont=font(15))
titulo = "Partícula Livre"*", "*L"$\sigma=$"*string(sigma)*", "* L"$dt=$"*string(dt)* ", " *L"$t=$" * string(round(t[i], digits=3))
plot!(title=titulo,
xlabel = "x",
xlim=(0,50),
xticksfont = font(13),
ticksfontsize = 10,
ylim=(0,1),
yticksfont=font(13),
)
i+=1
end
arquivo = "diracfree.gif"
gif(anim, arquivo, fps=30)
end
main()
Poço Infinito
O poço infinito foi simulado colocando dois potenciais muito grandes em e  . Abaixo está reproduzida a animação.
. Abaixo está reproduzida a animação.
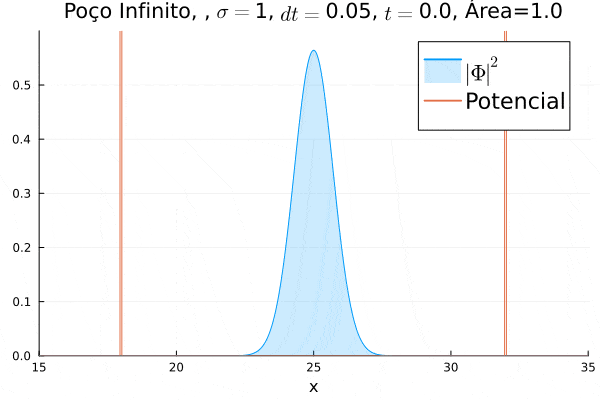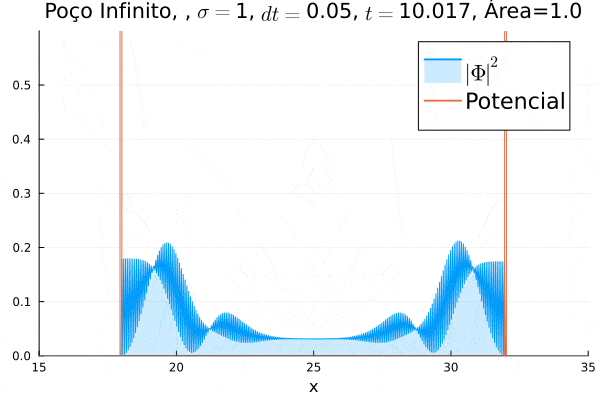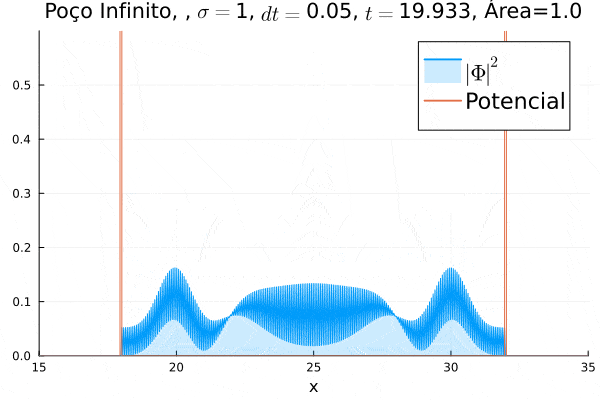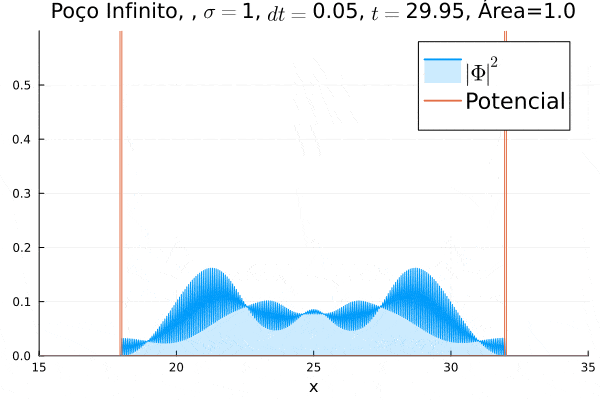
Nota-se que a função simulada é a soma dos quadrados dos módulos de cada função de onda (elétron e pósitron); é a integral de  que deve sempre ser unitária, o que concorda com o obtido. Na animação é possível perceber um comportamento semelhante ao de uma onda estacionária, onde tem-se vários "harmônicos" associados ao pacote de ondas: como esperado, a função de onda é uma combinação linear dos autoestados do poço infinito.
que deve sempre ser unitária, o que concorda com o obtido. Na animação é possível perceber um comportamento semelhante ao de uma onda estacionária, onde tem-se vários "harmônicos" associados ao pacote de ondas: como esperado, a função de onda é uma combinação linear dos autoestados do poço infinito.
Segue trecho do código utilizado para gerar a animação:
function main()
L = 50 #Dimensão espacial matriz
sigma = 1 #Largura do pacote gaussiano
dt = 0.05 #Passo temporal
size= 30*L #Utilizado para definir o passo espacial
h = L/size #Passo espacial
tmax = 30 #
tsize = round(Int64, tmax/dt) tamanho do vetor tempo.
t = LinRange(0, tmax, tsize)
x = LinRange(0, L, size)
V = zeros(size)
xi = init(LinRange(0, L, size), sigma) #Condição inicial de pacote gaussiano centralizado em x=25
#V = OHS(V,x,k)
V = poco(V, h) #Inicializa o potencial
f,g =CN(size,h,tsize, V, dt,xi) #Obtém as funções de onda e sua respectiva evolução temporal
##Produz-se a animação
i=1
anim = @animate while i<=tsize
f_real = real(f[i, :])
f_imag = imag(f[i, :])
g_real = real(g[i, :])
g_imag = imag(g[i, :])
probf = abs2.(f[i, :])
probg = abs2.(g[i, :])
prob = probf + probg
problem = SampledIntegralProblem(prob, x) #Resolve a integral em cada tempo, calculando a área e mostrando na animação
method = TrapezoidalRule()
integral = solve(problem, method)
integral = round(integral[1]; digits=3) #Arredonda.
integral = string(integral)
p = plot(x, prob,label=L"$|\Phi|^2$", legendfont=font(15), fill=true, fillalpha=0.2)
titulo = "Poço Infinito"*", "*", "*L"$\sigma=$"*string(sigma)*", "* L"$dt=$"*string(dt)* ", " *L"$t=$" * string(round(t[i], digits=3))*", Área="*integral
plot!(x,V, label="Potencial", legendfont=font(15))
plot!(title=titulo,
xlabel = "x",
xlim=(15,35),
xticksfont = font(13),
ticksfontsize = 10,
ylim=(0,0.6),
yticksfont=font(13),
)
i+=2 ##O passo é dado de 2 em 2 para deixar gif mais leve.
end
arquivo = "diracpoco.gif"
gif(anim, arquivo, fps=30)
end
main()
Oscilador Harmônico Simples
Inicialmente, realizou-se a simulação deslocando o pacote gaussiano para a posição inicial , com a finalidade de observar a oscilação. Testou-se a simulação com diferentes 's; escolheu-se o que está mostrado abaixo pois facilita a visualização do comportamento oscilatório.
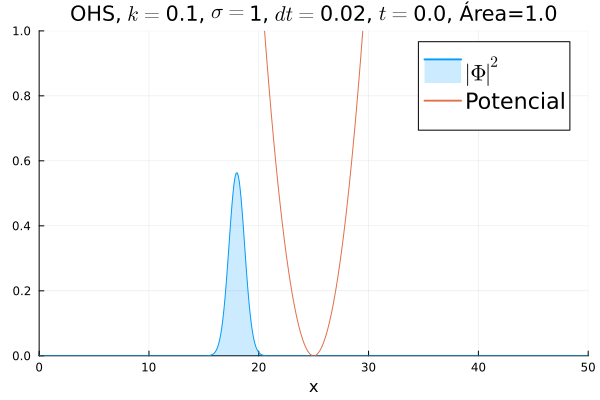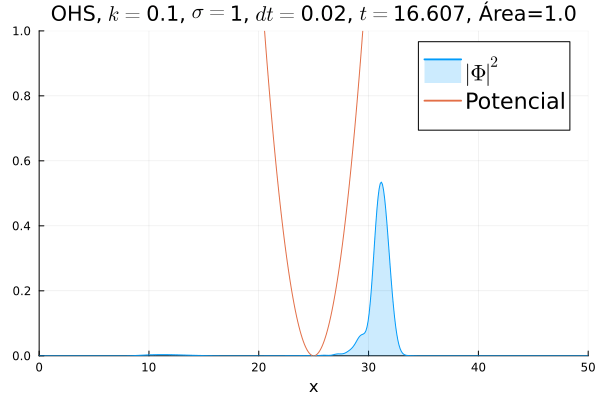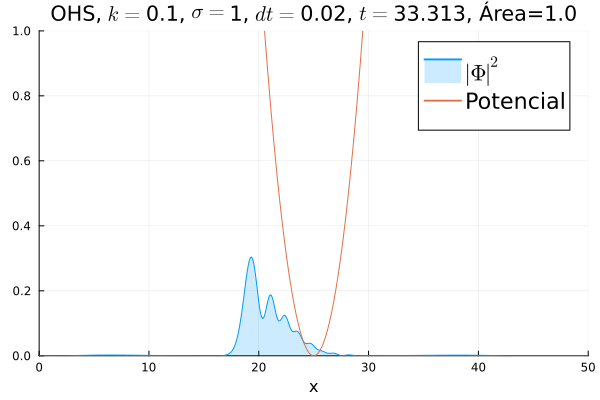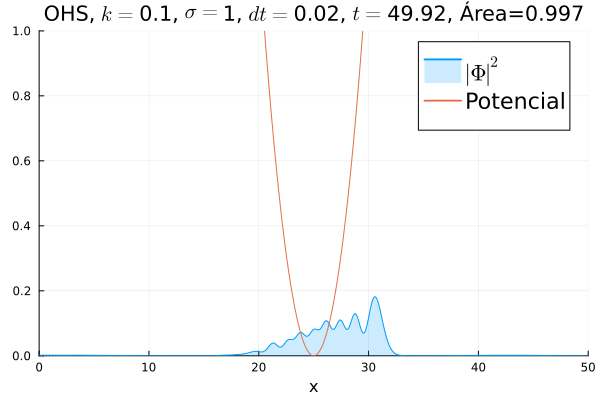
Nota-se um comportamento análogo ao caso clássico; porém, em alguns momentos é possível observar certos harmônicos do pacote de onda. Além disso, nota-se nas extremidades um valor máximo para  , também condizente com o caso clássico.
, também condizente com o caso clássico.
Durante testes nas simulações, notou-se que a área sob a curva possui variações quando se utiliza um passo muito alto. Nesse caso, a convergência do método de Crank-Nicolson para os valores teóricos depende da malha utilizada, embora a estabilidade dele seja incondicional. A diferença pode ser vista comparando-se a animação abaixo com a anterior:
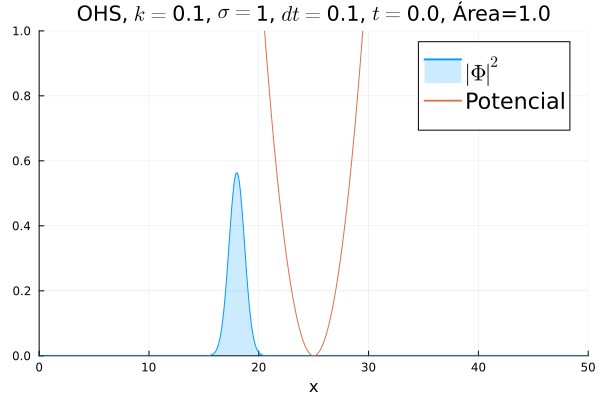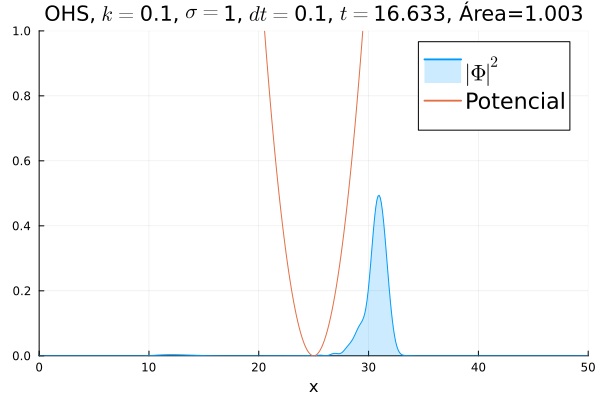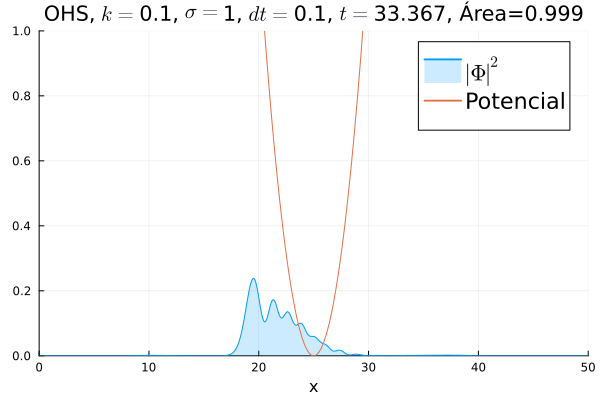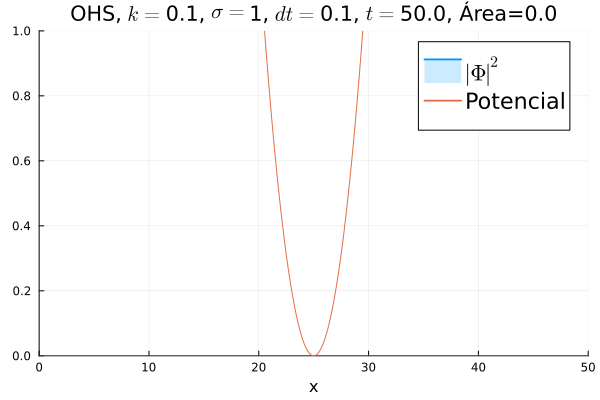
Em seguida, passou-se para uma condição inicial com um pacote gaussiano centralizado:
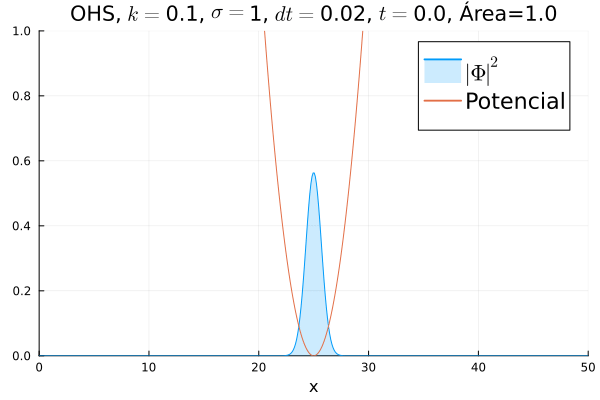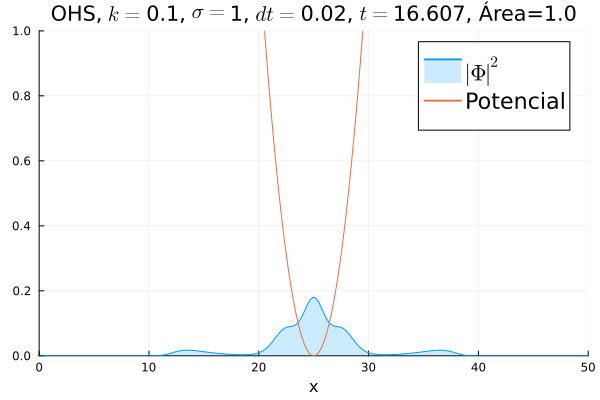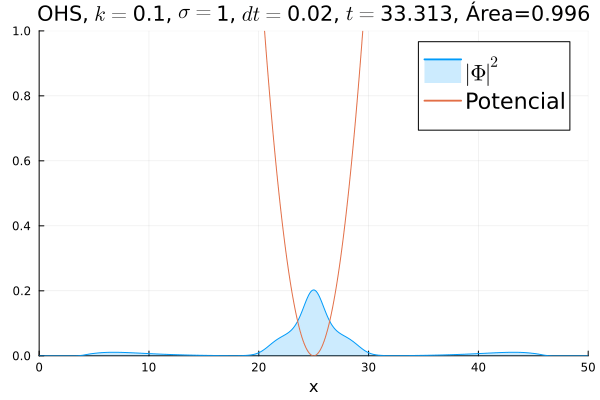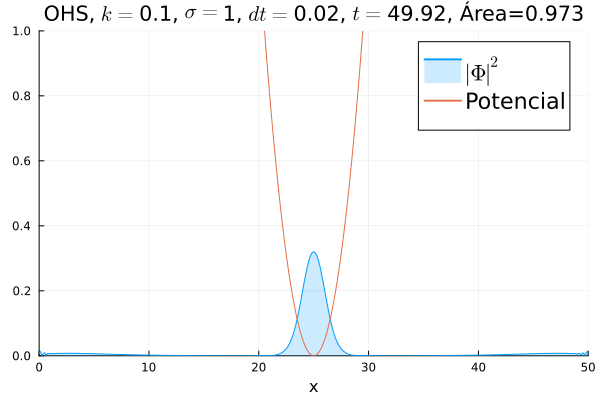
Mesmo que a posição inicial esteja no centro, devido ao fato da posição não estar bem definida ocorrem pequenas oscilações em torno do ponto de equilíbrio.
Por fim, segue o código utilizado para o oscilador harmônico simples (a diferença entre cada caso é só as constantes e a condição inicial).
function main()
L = 50 #Dimensão espacial matriz
k=0.1 #"Constante Elástica" do oscilador harmônico
sigma = 1 #Largura do pacote gaussiano
dt = 0.02
size= 10*L
h = L/size ##Passo espacial
tmax = 50
tsize = round(Int64, tmax/dt) #Tamanho do vetor tempo
t = LinRange(0, tmax, tsize)
x = LinRange(0, L, size)
V = zeros(size)
xi = init(LinRange(0, L, size), sigma) #Condição inicial pacote gaussiano
V = OHS(V,x,k) ##inicializa o potencial
f,g =CN(size,h,tsize, V, dt,xi)
i=1
#Faz o gif
anim = @animate while i<=tsize
f_real = real(f[i, :])
f_imag = imag(f[i, :])
g_real = real(g[i, :])
g_imag = imag(g[i, :])
probf = abs2.(f[i, :])
probg = abs2.(g[i, :])
prob = probf + probg
problem = SampledIntegralProblem(prob, x) ##Calcula a área sob a curva
method = TrapezoidalRule()
integral = solve(problem, method)
integral = round(integral[1]; digits=3)
integral = string(integral)
p = plot(x, prob,label=L"$|\Phi|^2$", legendfont=font(15), fill=true, fillalpha=0.2)
#=
p=plot(x, probf, label=L"$|\phi_1|^2$", legendfont=font(15))
plot!(x, probg, label=L"$|\phi_4|^2$", legendfont=font(15))
=#
titulo = "OHS"*", "*L"$k=$"*string(k)*", "*L"$\sigma=$"*string(sigma)*", "* L"$dt=$"*string(dt)* ", " *L"$t=$" * string(round(t[i], digits=3))*", Área="*integral
plot!(x,V, label="Potencial", legendfont=font(15))
plot!(title=titulo,
xlabel = "x",
xlim=(0,50),
xticksfont = font(13),
ticksfontsize = 10,
ylim=(0,1),
yticksfont=font(13),
)
i+=5 ##Feito de 5 em 5 para deixar .gif mais leve
end
arquivo = "diracOHS.gif"
gif(anim, arquivo, fps=30)
end
main()
Referências
- The quantum theory of the electron. Proceedings of the Royal Society of London A, v. 117, n. 778, p. 610–624, fev. 1928.
- SAKURAI, J. J. Mecânica quântica moderna. 2. ed. Porto Alegre: Bookman, 2012.
- BAO, W. et al. Numerical Methods and Comparison for the Dirac Equation in the Nonrelativistic Limit Regime. Journal of Scientific Computing, v. 71, n. 3, p. 1094–1134, jun. 2017.
- SOFF, G. et al. Solution of the Dirac Equation for Scalar Potentials and its Implications in Atomic Physics. Zeitschrift für Naturforschung A, v. 28, n. 9, p. 1389–1396, 1 set. 1973.
- THALLER, B. The Dirac equation. Berlin: Springer, 2010.




