Simulação de Enovelamento de Proteína
Carolina Lenzi
Este trabalho tem como objetivo utilizar o método de Monte Carlo para simular o enovelamento de proteínas.
Proteínas
As proteínas são moléculas com papel fundamental para os seres vivos. Elas atuam em diversos processos biológicos, como o transporte de oxigênio, a catálise de reações químicas, a defesa do organismo, a formação dos hormônios e formação de ossos e tendões. As proteínas são formadas por centenas de aminoácidos ligados em cadeia por ligações covalentes. Existem 20 aminoácidos proteinogênicos que dão origem às proteínas conhecidas e encontradas na natureza. Cada sequência de aminoácidos ligados covalentemente gera uma proteína diferente, com uma função específica no organismo. Essa sequência, bem determinada, é chamada de estrutura primária da proteína.

A proteína tende a se enovelar para minimizar a energia da sua estrutura, relativa às interações não covalentes (como ligações de hidrogênio, ponte salina, interações hidrofóbicas, interações iônicas…) entre os aminoácidos e com o meio. Isso é possível porque as ligações químicas possuem uma certa flexibilidade, que permite a rotação das moléculas. O enovelamento da estrutura primária gera regiões com padrões regulares na proteína, chamadas de estrutura secundária. Essas regiões regulares também tendem a se enovelar, dando origem a estrutura terciária da proteína. A estrutura terciária de menor energia é, geralmente, a conformação funcional da proteína, ou seja, sua configuração em estado biologicamente ativo, que é conhecida como forma nativa. Em alguns casos as estruturas terciárias podem se aglomerar e formar as estruturas quaternárias.
Como a função da proteína está diretamente relacionada com a sua forma nativa, e esta está relacionada com a sequência de aminoácidos que dá origem à proteína, é interessante conseguir prever a estrutura terciária a partir da estrutura primária, com a finalidade de desenvolver proteínas com características específicas. Esse é um dos grandes problemas da bioinformática atualmente. Além da complexidade de compreender a termodinâmica e o mecanismo do enovelamento, a proteína encontra a estrutura ótima em fração de segundo, mas o tempo que seria necessário para testar aleatoriamente as estruturas possíveis para encontrar a de menor energia é maior que a idade do universo, como apontado no paradoxo de Levinthal.
Simulação de Monte Carlo
A simulação do enovelamento foi feita com base no livro do Giordano e envolve um modelo muito simplificado do que acontece na realidade. O código foi implementado em C e os gráficos foram gerados no gnuplot.
Consideramos uma cadeia de N aminoácidos, sorteados dentre os 20 possíveis, para montar a estrutura primária da proteína, representada por um vetor de tamanho N. Aminoácidos em posições adjacentes do vetor são considerados ligados covalentemente. A cadeia é colocada em uma rede quadrada de tamanho NxN, para permitir que a proteína esteja completamente desenovelada (esticada). Em cada posição da rede, um aminoácido tem no máximo 4 aminoácidos vizinhos mais próximos com os quais pode interagir ou estar ligado, sendo eles acima, abaixo, à direita e à esquerda. Para as interações de estabilização da proteína, consideramos as seguintes forças atuantes:


- Forças de van der Waals entre aminoácidos não ligados covalentemente: força atrativa para aminoácidos próximos, que perde o efeito com o aumento da distancia entre os aminoacidos;
- Ligações de hidrogênio: ligações entre aminoácidos próximos na rede;
- Interação com a água (hidrofilicidade e hidrofobia): aminoácidos hidrofílicos são atraídos pela água presente no meio e, por isso, tendem a manter a cadeia não enovelada. Já os hidrofóbicos repelem a água do meio e preferem uma estrutura enovelada.
Todas essas forças estão competindo no processo de enovelamento da proteína. Para o modelo, agrupamos essas interações em uma energia , associada a um par de aminoácidos vizinhos na rede, A(m) e A(n) nas posições i e j, e não ligados covalentemente, ou seja, não adjacentes na cadeia. A energia da estrutura é dada pela soma sobre todos os pares de aminoácidos da proteína:
onde se os aminoácidos m e n são vizinhos na rede e não estão ligados covalentemente, e zero caso contrário.
No livro, os autores sugerem pensar as energias como uma matriz 20x20 contendo todas as interações entre os 20 aminoácidos considerados. Porém, como a interação depende apenas de quais são os aminoácidos do par, a matriz é simétrica , e podemos guardar apenas 210 valores correspondentes aos pares distintos de aminoácidos.
Com essas definições, podemos partir para a simulação de Monte Carlo. Inicialmente, sorteamos um aminoácido e encontramos a posição dele na rede. Cada posição tem no máximo 8 vizinhos para onde o aminoácido pode (ou não) ser movido, que são os 4 vizinhos mais próximos e as diagonais para cima e para baixo. Calculamos a energia inicial da estrutura e partimos para a tentativa de movimento: sorteamos um de seus vizinhos e analisamos se é possível mover o aminoácido. Primeiro, verificamos se a rede está vazia na posição do vizinho sorteado e, em caso positivo, verificamos se nenhuma ligação entre aminoácidos será comprimida ou esticada com o movimento. Se essas duas condições forem satisfeitas, realizamos o movimento e calculamos a energia final da estrutura. Se a energia final for menor que a inicial, aceitamos o movimento e voltamos ao primeiro passo. Se a energia final for maior que a inicial, aceitamos o movimento apenas se o fator de Boltzmann for maior que um valor aleatório entre 0 e 1. Caso o movimento não seja aceito, recuperamos a estrutura inicial da proteína. Repetimos esses passos diversas vezes e, a cada tentativa de movimento, contamos um tempo de Monte Carlo.
Resultados

Em todas as simulações, a posição inicial da cadeia foi esticada horizontalmente no meio da matriz da rede. Os valores de foram sorteados dentro do intervalo [-4, -2], como utilizado no livro. As energias foram medidas em unidades de kB por simplicidade.
Na figura 1, observamos o gráfico da energia em função do tempo durante a simulação para uma proteína com 15 aminoácidos à temperatura T = 10. As flutuações de energia são grandes pois a temperatura é alta, o que aumenta a probabilidade de alterar a estrutura da proteína. Já na figura 2, temos também a energia em função do tempo para uma proteína com 15 aminoácidos, mas à temperatura T = 1. Nesse caso, as flutuações diminuem consideravelmente e a proteína passa a maior parte do tempo em estados de menor energia.
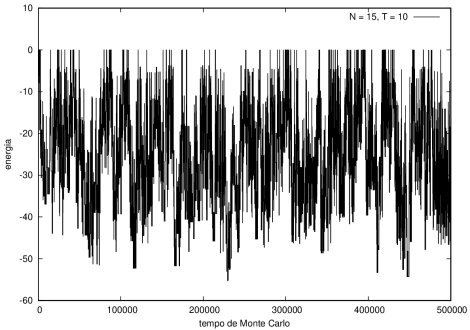
Figura 1 Energia em função do tempo para proteína com 15 aminoácidos. T = 10 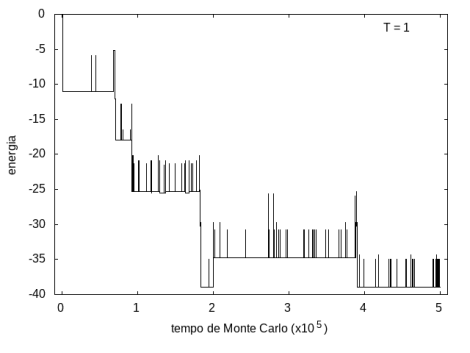
Figura 2 Energia em função do tempo para proteína com 15 aminoácidos. T = 1 
Figura 7 Estrutura da proteína com 15 aminoácidos após 2e5 tempo de monte carlo, T = 10



Nas figuras a seguir, a mesma comparação anterior, mas para o caso de proteínas com 30 (figuras 3 e 4) e com 100 (figuras 5 e 6) aminoácidos.
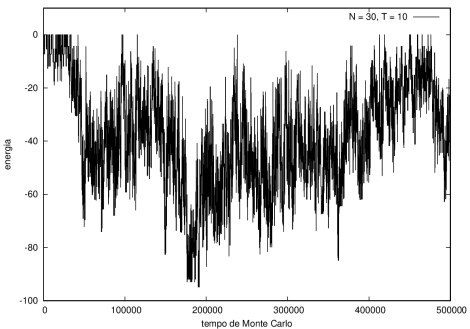
Figura 3 Energia em função do tempo para proteína com 30 aminoácidos. T = 10 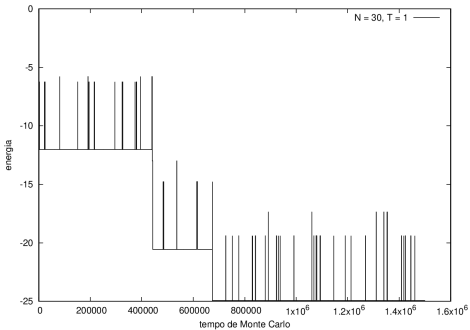
Figura 4 Energia em função do tempo para proteína com 30 aminoácidos. T = 1 
Figura 8 Estrutura da proteína mesma proteína da figura 7 após 5e5 tempo de monte carlo, T = 10



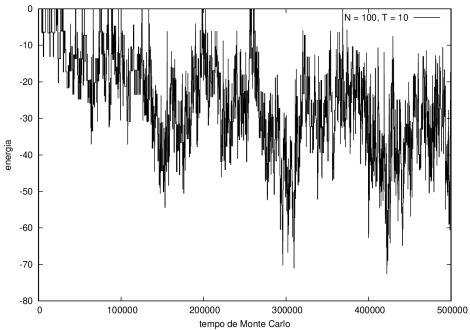
Figura 5 Energia em função do tempo para proteína com 100 aminoácidos. T = 10 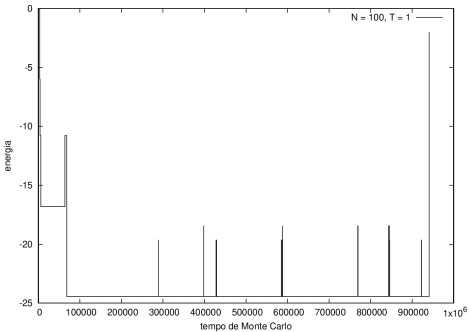
Figura 6 Energia em função do tempo para proteína com 100 aminoácidos. T = 1


Abaixo, duas configurações da mesma proteína com 30 aminoácidos durante a simulação, para T = 1.

Na figura a seguir, duas estruturas da mesma proteína com 100 aminoácidos durante a simulação, com temperatura T = 10.
